Instructions for PeptideMass Peptide Characterisation Software
1. Introduction:
This program is designed to calculate the theoretical masses of peptides generated by the chemical or enzymatic cleavage of proteins, to assist in the interpretation of peptide mass fingerprinting and peptide mapping experiments. Protein sequences can be provided by the user or can be a code name for a protein in the UniProt Knowledgebase (Swiss-Prot or TrEMBL). When proteins of interest are specified from UniProtKB/Swiss-Prot, the program considers all annotations for that protein in the UniProtKB/Swiss-Prot database, and uses these in order to generate the correct peptide masses and warn users about peptides that are not likely to be found when undertaking peptide mass fingerprinting. Many protein post-translational modifications which affect the masses of peptides can thus be taken into consideration.2. Using PeptideMass:
In the program there are a number of fields that must be filled in. Here we will describe these fields, from the top to the bottom of the form.
In the following set of notes, instructions you should follow for filling in
the form on the computer will be shown with a  . Background notes relevant
to that part of the form will be marked with a
. Background notes relevant
to that part of the form will be marked with a 
2.1. Protein Sequence to be Cleaved
Here you should specify the sequence of the protein you would like to
cleave. If this protein is known in UniProtKB, enter the
UniProtKB/Swiss-Prot ID code (e.g. ALBU_HUMAN) or the protein accession number (e.g. P02769).
If the protein
is not known in UniProtKB, you can enter the sequence of your
protein of interest, in single letter amino acid code, in either upper or
lower case. However, you can only specify one sequence at a time in this case.
2.2. Enzyme or Reagent to Use for Cleavage
Here you should select the enzyme or reagent with which you would like to
program to use in calculating the theoretical peptide masses. The rules
that are used to cleave the proteins are shown in Table 1.
2.3. Modifications to Cysteines in Peptides
You can choose how you would like all cysteines in a protein to be
modified, before the theoretical masses of peptides are calculated.
Experimentally, proteins are usually subjected to reduction and then
alkylation with different reagents before they are used to generate
peptides. If you would like the masses of unmodified cysteines in your
peptides, check "nothing (in reduced form)" in the menu 'cysteines treated with'. If you would like all
cysteines to be theoretically reduced and alkylated, specify
the reagent to be used for alkylation.
You have a choice of iodoacetamide, iodoacetic acid and 4-vinyl pyridene.
If you wish to generate masses for peptides with acrylamide adducts, you can check
the box 'with acrylamide adducts'.
The program will then modify the theoretical masses of
Cys-containing peptides accordingly. Note that in proteins prepared by
polyacrylamide gel electrophoresis, it can be common for cysteines to have
reacted with free acrylamide monomers.
2.4. Modifications to Methionines in Peptides
You can request for all methionines in theoretical peptides to be
oxidised. If this option is selected, the program will modify the
theoretical masses of all Met-containing peptides accordingly. Note that
proteins prepared by gel electrophoresis often show this modification.
2.5. Peptide Mass Range & Sorting
Here you can specify a low mass cutoff, such that any peptides below that
mass will not be shown in the results. You should then choose if you would
like the peptides to be sorted by their mass (from largest to smallest) or
by their chronological order in the protein.
2.6. Modifications, Conflicts, Variants, and Alternative Splicing
Here you can specify which of the annotations in the UniProtKB/Swiss-Prot database
you would like to be taken into account, or warned of, during the
calculation of your peptide masses. If you select any of these, the program
will print them in the results output if they are known for your protein of
interest.
2.7. Reset and Perform Buttons
Once you have filled in the form according to your needs, press the
Perform button. If you have made a mistake and would like all fields to be
reset to their default values, press the Reset button.
3. Special Features of PEPTIDE MASS:
The following section provides details as to how the program works, and how it uses a maximum of information available from the UniProtKB/Swiss-Prot database to calcluate the masses of peptides.3.1. Mass Calculations
Masses have been calculated to 4 or 5 decimal places for all amino acids and post-translational modifications. Average isotopic and monoisotopic mass values are available for all of these modifications.3.2. Enzymes
The rules that are used to cut proteins in the program are as summarised in Table 1. Note that to take into account partial cleavages, it is possible to specify a maximum number (0, 1, 2, or 3) of missed cleavages to be considered.Table 1: Cleavage rules for PEPTIDE MASS program.
| Enzyme or Reagent | Cleaves where? | Exceptions |
|---|---|---|
| Trypsin | C-terminal side of K or R | if P is C-term to K or R |
| Trypsin (C-term to K/R, even before P) | C-terminal side of K or R | |
| Trypsin (higher specificity) | C-terminal side of K or R | if P is C-term to K or R; after K in CKY, DKD, CKH, CKD, KKR; after R in RRH, RRR, CRK, DRD, RRF, KRR |
| Lys C | C-terminal side of K | |
| Lys N | N-terminal side of K | |
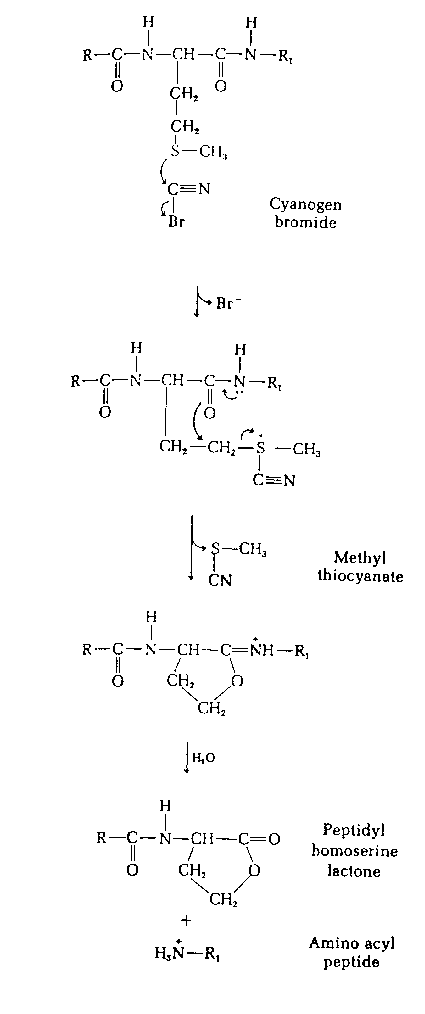
| CNBr | C-terminal side of M | |
| Arg C | C-terminal side of R | if P is C-term to R |
| Asp N | N-terminal side of D | |
| Asp N + Lys C | N-terminal side of D, C-terminal side of K | |
| Asp N + N-terminal Glu | N-terminal side of D or E | |
| Asp N + Glu C (bicarbonate) | N-terminal side of D, C-terminal side of E | |
| Glu C (bicarbonate) | C-terminal side of E | if P is C-term to E, or if E is C-term to E |
| Glu C (phosphate) | C-terminal side of D or E | if P is C-term to D or E, or if E is C-term to D or E |
| Glu C (phosphate) + Lys C | C-terminal side of D, E and K | if P is C-term to D or E, or if E is C-term to D or E |
| Glu C (phosphate) + Trypsin | C-terminal side of D, E, K, R | if P is C-term to K, R, D, E, or if E is C-term to D or E |
| Glu C (phosphate) + Chymotrypsin | C-terminal side of F, L, M, W, Y, D, E | if P is C-term to F, L, M, W, Y, D, E, or if E is C-term to D or E, not after Y if P is N-term to Y |
| Microwave-assisted formic acid hydrolysis (C-term to D) | C-terminal side of D | |
| Chymotrypsin (C-term to F/Y/W/M/L, not before P, not after Y if P is C-term to Y) | C-terminal side of F, L, M, W, Y | if P is C-term to F, L, M, W, Y, if P is N-term to Y |
| Chymotrypsin (C-term to F/Y/W/, not before P, not after Y if P is C-term to Y) | C-terminal side of F, Y, W | if P is C-term to F, Y, W, if P is N-term to Y |
| Trypsin/Chymotrypsin (C-term to K/R/F/Y/W, not before P, not after Y if P is C-term to Y) | C-terminal side of K, R, F, Y, W | if P is C-term to K, R, F, Y, W, if P is N-term to Y |
| Pepsin (pH 1.3) | C-terminal side of F, L | |
| Pepsin (pH > 2) | C-terminal side of F, L, W, Y, A, E, Q | |
| Proteinase K | C-terminal side of A, F, Y, W, L, I, V | |
| Thermolysin | N-terminal side of A, F, I, L, M, V | if D or E is N-term to A, F, I, L, M, V |
{kind=link}
3.3. Signal Sequences, Propeptides & Transit Peptides
Signal sequences, propeptides and transit sequences are all removed from proteins before cleavage rules are applied. A message is shown at the top of the list of results if any of these are present in a protein. All will be removed before generating the masses of peptides from the mature protein.3.4. Chains and Polypeptides that Produce Multiple Mature Proteins
If there are known chains that are created from any database entry, these are considered as different polypeptides (e.g. A2HS_HUMAN). Thus in the list of results, there will be a different list of peptides for each of these chains. The same applies to any proteins which are known to form multiple mature peptides or proteins from a single initial polypeptide (e.g. COLI_HUMAN).3.5. Protein Post-Translational Modifications
All documented post-translational modifications of a protein in the UniProtKB/Swiss-Prot database (including the annotations MOD_RES, LIPID, CARBOHYD, and DISULFID), are considered by the program. It will indicate the peptide which will carry the modification, detailing the type of modification and the number of the residue that carries it. The type of the modification is usually represented by the first four letters of the code used in UniProtKB/Swiss-Prot for that post-translational modification.For modifications that are simple and discrete (acetylation, amidation, biotin, C-mannosylation, deamidation, dimethylation, farnesylation, formylation, geranyl-geranyl, gamma-carboxyglutamic acid, O-GlcNac, hydroxylation, methylation, myristoylation, palmitoylation, phosphorylation, pyrrolidone carboxylic acid, sulfatation and trimethylation), the predicted modified mass of the peptide will be given. Currently, if there is more than one type of modification in any peptide, a separate mass of the peptide will be given for each type of modification. Thus there can be more than one modified mass for a single peptide. For complex post-translational modifications, including N- and O- glycosylation and phosphatidyl inositol glycan anchors, it is difficult or impossible to predict the modified mass of a peptide. No predicted theoretical mass is supplied in such cases.
3.6. Conflicts in the Database
If there are known conflicts in the database, which may represent database errors, these are shown under the "conflict" column, corresponding to the peptide that may be affected. In effect this is a caution that such peptides may not necessarily be found in preparations of a sample. It is currently not possible (apart from manually modifying the sequence and re-cutting with the program) for the program to give you the masses of any alternative peptides that would be created by the conflict. However, to facilitate further investigation of such cases, the conflict itself is shown in the "conflict" column (e.g. 168: F -> K). If there is more than one residue changed in any peptide, the number represents the number of the first changed residue. If there is a residue conflict that is a missing amino acid, it will be represented as, for example: 168-169 MISS.Refer to the user manual to see how conflicts are annotated in UniProtKB/Swiss-Prot.
3.7. Variants in the Database
If there are known variants in the database, representing isoforms of the same protein, these will be shown corresponding to the peptide that may be affected. To facilitate further investigation of such cases, the variant itself is shown in the "variant" column (e.g. 46: L -> S). If there is more than one residue changed in any peptide, the number represents the number of the first changed residue (e.g. 48: GLVVR -> PSSCARV).Refer to the user manual to see how variants are annotated in UniProtKB/Swiss-Prot.
3.8. Alternative Splicing in the Database
If there are known protein isoforms which correspond to differentially spliced versions of a single mRNA species (e.g Q02040), these will be shown in the "varsplice" column corresponding to the peptide that may be affected. The program will show the numbers of the residues that may be affected in the peptide, by either the addition of extra residues, or by the deletion of some residues from that area. The letter code of the amino acids within any particular affected peptide are not shown. If you wish to perform a theoretical digest of one of the annotated splice isoforms, you can click on the IsoId in the NiceProt view of the underlying UniProtKB entry, e.g. the short isoform Q02040-2 in Q02040. This leads to a page displaying the sequence of this isoform, and containing links for the direct submission of that sequence to a number of tools, including Peptide-Mass.Refer to the UniProtKB entry view manual to see how alternative splicing is annotated in UniProtKB/Swiss-Prot.
3.9. Hypertext Link in Results Page
If a protein from the UniProtKB is the protein under study, a hypertext link will be provided to the protein at the top of the results page. This link allows the user to effortlessly retrieve the full UniProtKB listing for that protein. Note that if the user wishes to, for example, change the sequence from a UniProtKB/Swiss-Prot entry to take into account a variant, it is easy for the sequence to be copied from the UniProtKB/Swiss-Prot entry, pasted into the sequence field (see 2.1.), modified as necessary, and then cleaved as required.In case of known chains or multiple mature peptides (see 3.4.), hypertext links are provided to a page that highlights the subsequence in question.