NOMAD V1.0
About
Nomad (Neighborhood Optimization for Multiple Alignment Discovery) is a program dedicated to the ungapped local multiple alignment (ULMA) problem, also known as "blocks". By using an entropy-based objective function that takes into account the amino acid's nature, Nomad is well suited to deal with protein sequences. This objective function, the shared entropy, has been shown to be significantly more reliable than the relative entropy when protein sequences to be aligned are distantly related.References
- Hernandez D. Gras R. Appel R. Neighborhood Function and Hill-Climbing Strategies dedicated to the Generalized Ungapped Local Multiple Alignment. Eur J Oper Res, 2006, in press (doi:10.1016/j.ejor.2005.10.076).
- Hernandez D. (2005) Stratégies d'optimisation combinatoire pour le problème de l'alignement local multiple sans indels, et application aux séquences protéiques. PhD thesis, Université de Genève, SWITZERLAND.
Overview
An ULMA is essentially a collection of n occurrences of size w , chosen in way to be maximally conserved. Both n and w are fixed by the user. Nomad is an optimization program that makes use of a hill-climber to search the n occurrences that maximize an objective function.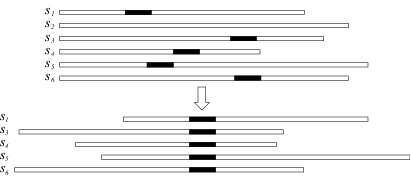
(a) OOPS (One Occurrence Per Sequence)
This is the simplest and the most constrained mode. It is supposed that every sequence contributes exactly once to the ULMA. In this mode, n is implicitly fixed by the number of sequences in the dataset.
(b) ALOOPS (At Least One Occurrence Per Sequence)
All sequences must contribute to the ULMA but some may contribute more than once. n has to be specified as greater than or equal to the number of sequences.
(c) AMOOPS (At Most One Occurrence Per Sequence)
Some sequences can be discarded from the ULMA. n has to be specified as lower than or equal to the number of sequences.
(d) AOPS (Any number of Occurrences Per Sequence)
This mode is the least constrained one. It allows occurrences to be distributed anywhere in the sequence set, as long as they do not overlap with each other. n has to be specified between 2 and a reasonable value.

The widely used objective function for the ULMA problem is the relative entropy, which is the information theory point of view of a log-likelihood ratio statistic. The main drawback of the relative entropy when aligning protein sequences is that all amino acids are considered to be independent. The fact that some substitutions may occur more often than others is not considered by this function. Nomad implement the "shared entropy", an objective function which takes into account an "equivalence" measure between amino acids. The shared entropy has been shown to be significantly more efficient than the relative entropy, both in terms of noise/signal distinction, and optimization process.
Input
Dataset:
Paste your sequences in the FASTA format.
Example:
>sequence label
MKALTARQQEVFD...
>sequence label
MEQNPQSQLKLLV...
>sequence label
MGMKISELAKACD...
Width:
Set the width of the ULMA to be searched.
Protein, shared entropy:
This is the default option. The ULMA is optimized with the shared entropy objective function. This option is highly recommended for protein sequences, especially when they are distantly related.
Protein, relative entropy:
Optimize the ULMA with the "classical" relative entropy objective function. The relative entropy is the widely used function for the ULMA problem.
DNA, relative entropy:
Choose this option if you align DNA sequences.
Occurrence repartition constraints:
Choose one of the OOPS, ALOOPS, AMOOPS or AOPS constrain models and set the number of occurrences in the ULMA.
Sort occurrences:
Check this option to sort occurrences by their own score. The score of an occurrence is a log-likelihood ratio, which reflects how well the occurrence fits the rest of the ULMA.
E-mail address:
Type your e-mail address to get the result in your mail box. This option is recommended and is more reliable if the cpu-time is substantial.
Explanation of the result
This example shows a ULMA under the OOPS mode performed on 15 helix-turn-helix domain-containing proteins. The first column shows the label of the sequence, the second column gives the occurrence positions in the corresponding sequence. The third column shows the occurrence itself, and finally the fourth column shows the score of the occurrence. This score reflects how well the occurrence fits the rest of the alignment. The alignment score is the objective value that has been optimized, and correspond to the average occurrence score. Note that these scores cannot be interpreted as confidence values. They are only relative to the ULMA that has been optimized and thus cannot been compared between different ULMAs. Symbols are blue-scaled according to their objective score contribution. The darker the symbol the stronger its contribution.Since Nomad performs stochastic optimizations, two independent runs with the same parameters could produce a different result. If this occurs, simply consider the best scoring alignment.
>LEXA_ECOLI_P03033; 26 P T R A E I A Q R L G F R S P N A A E E H L 15.691
>RPSD_ECOLI_P00579; 571 Y T L E E V G K Q F D V T R E R I R Q I E A 19.645
>MERR_STAAU_P22874; 3 M K I S E L A K A C D V N K E T V R Y Y E R 19.422
>ASNC_ECOLI_P03809; 23 T A Y A E L A K Q F G V S P G T I H V R V E 22.185
>ICLR_ECOLI_P16528; 44 V A L T E L A Q Q A G L P N S T T H R L L T 18.815
>LACR_STAAW_P16644; 20 I R T N E I V E G L N V S D M T V R R D L I 16.389
>CRP_ECOLI_P03020; 168 I T R Q E I G Q I V G C S R E T V G R I L K 20.827
>GNTR_BACLI_P46833; 42 L S E N K L A A E F S V S R S P I R E A L K 17.506
>PMX1_MOUSE_P43271; 122 F V R E D L A R R V N L T E A R V Q V W F Q 18.347
>LYSR_ECOLI_P03030; 19 G S L T E A A H L L H T S Q P T V S R E L A 18.060
>ARSR_STAAU_P30338; 30 L C A C D L L E H F Q F S Q P T L S H H M K 20.778
>ARAC_ECOLI_P03021; 195 F D I A S V A Q H V C L S P S R L S H L F R 19.659
>NER_BPMU_P06020; 23 L S L S A L S R Q F G Y A P T T L A N A L E 19.644
>RCRO_BPP22_P09964; 11 G T Q R A V A K A L G I S D A A V S Q W K E 18.936
>FIXJ_BRAJA_P23221; 158 L S N K L I A R E Y D I S P R T I E V Y R A 17.702
Objective score 18.907