GlycoMod - Documentation
GlycoMod is a program designed to find all possible compositions of a glycan structure from its experimentally determined mass. This is done by comparing the mass of the glycan to a list of pre-computed masses of glycan compositions.
The program can be used with free or derivatized glycans and for glycopeptides where the peptide mass or protein is known. Compositional constraints can be applied to the output.
The program can be used with free or derivatized glycans and for glycopeptides where the peptide mass or protein is known. Compositional constraints can be applied to the output.
Input parameters - Output - Credits
Input parameters
-
Experimental masses
The user may enter the experimental masses to be analyzed, separating them by spaces or new lines. It is also possible to enter the masses from a text file provided each mass is on a new line. These mass values may be average or monoisotopic, but the user must select the appropriate button, and the mass values must all be in agreement. A mass tolerance level should be selected in either Daltons or ppm. Note that the higher the mass tolerance, the greater the number of compositions returned.
The experimental masses may correspond to glycopeptides or free oligosaccharides, which may be derivatized (see below).
-
Ion mode and adducts
The user may enter the masses as neutral ions, positive ions, or as negative ions. Examples of these are [M], [M+H]+, [M+Na]+, [M+K]+, [M+H]-, [M+CH3COO]- or [M+TFA]-.
-
Glycan form
GlycoMod can calculate the possible compositions of N-linked oligosaccharides, linked via the amide nitrogen of an asparagine residue, or O-linked oliogsaccharides, linked via the hydroxyl group of serine or threonine. [Note: Oligosaccharides may also be O-glycosidically linked via the hydroxyl group of hydroxylsine, hydoxyproline and tyrosine. These amino acid linkages are less common and are not considered in this version of GlycoMod.]
GlycoMod can calculate the composition of the glycans from the masses of glycopeptides or of glycans released from the peptide moiety by enzymatic or chemical means.
-
Glycopeptides
GlycoMod may be used to calculate the possible composition of a glycan on a glycopeptide. The peptide data may be entered as a protein sequence, a UniProtKB AC or ID, or as a set of unmodified peptide masses ([M], where the masses are average or monoisotopic in agreement with that specified for the experimental masses of the data entered above).
When a UniProtKB AC or ID is entered, the protein may be digested with a number of enzymes. These are: Trypsin
Lys C
Arg C
Asp N
Asp N + N-terminal Glu
Glu C in a bicarbonate buffer
Glu C in a phospate buffer
Glu C in a phospate buffer + Lys C
Chymotrypsin.
The digest may also be performed using CNBr.
The cleavage rules of all these enzymes can be looked up in the PeptideMass documentation.
In all cases the user may select 0, 1, 2, or 3 missed cleavage sites.
It is possible to choose how the cysteines in a protein might be modified. For example, many researchers subject their proteins to reduction and alkylation of the cysteines with a variety of reagents to aid in enzyme digestion for the generation of peptides. In GlycoMod it is possible to select the cysteines as unmodified (the default value), or as reduced and alkylated using
- iodoacetic acid - caboxymethyl cysteine, Cys-CM
- iodoacetamide - carboxyamidomethyl cysteine, Cys_CAM
- 4-vinyl pyridene - pyridyl-ethyl cysteine, Cys_PE
It is also possible to select acrylamide adducts on cysteines. This is a common occurrence when proteins are prepared using polyacrylamide gel electrophoresis.
When a cysteine modification has been selected, GlycoMod considers peptides with both unmodified and modified cysteines. If more than one cysteine residue occurs in a peptide, the masses of all possible combinations of modified and unmodified residues is calculated. For example, if a peptide contains 3 cysteine residues then GlycoMod considers the masses for that peptide containing 0, 1, 2 and 3 modified residues.
Another common modification when proteins are prepared using polyacrylamide gel electrophoresis is that the methionines in a peptide are oxidised. If this option is selected, the program will modify the theoretical masses of Met-containing peptides accordingly and consider both peptides with unmodified methionines and peptides with modified methionines, in the same manner as for modified cysteines.
When a protein sequence or a UniProtKB ID or AC is entered, GlycoMod only considers those peptides that have the sequence NX[STC] where X≠Pro for N-linked glycans, and peptides that contain S and/or T for O-linked glycans. Where there are multiple sites, e.g. in mucin glycopeptides, GlycoMod calculates the glycan composition as if there was only one site. Therefore, the glycan composition given may actually consist of more than one glycan structure. This is also true where there is heterogeneity in the glycan structures present on one amino acid.
-
Released glycans
GlycoMod may be used to calculate the possible composition of a glycan from its mass after its release from a protein or peptide. The form of the glycan moiety may be as a free, reduced or derivatized glycan.
N-linked oligosaccharides are described as free when released using PNGase F, PNGase A or released by anhydrous hydrazine and regenerated to reducing oligosaccharides. Endo H and Endo F released N-linked glycans are considered separately, due to the fact that these enzymes cleave the GlcNAc(β1-4)GlcNAc core linkage, thereby resulting in one less GlcNAc moiety remaining in the glycan composition.
Similarly O-linked glycans may be described as free oligosaccharides if released using O-glycanase, or by mild hydrazinolysis followed by regeneration to the reducing oligosaccharides, or by non-reductive beta-elimination.
To prevent base degradation (“peeling”) O-linked oligosaccharides are traditionally released by the popular reductive beta-elimination method. This method releases the oligosaccharides and reduces them to alditols, i.e. reduced oligosaccharides.
Once released, free reducing oligosaccharides are often derivatized at the reducing terminus by a process of reductive amination, i.e. the reducing terminus of the glycan is reacted with an amine followed by reduction with a selective reducing agent. Common derivatives include 2-aminopyridine (PA), 2-aminobenzoic acid (ABA) or 8-aminonapthalene-1,3,6-trisulfonic acid (ANTS).
When “Derivatized oligosaccharides” is chosen, it is essential for the user to identify the derivative and to supply its mass [M] in the appropriate boxes labeled “derivative” and “mass” located further down the form. The mass required is the monoisotopic or average mass of the non-reacted derivative, e.g. 94.053 for the monoisotopic mass of 2-aminopyridine (PA). The calculation for the addition of a derivative automatically adds the mass of 2 hydrogen atoms. These are automatically added for the addition of a hydrogen atom to the non-reducing terminus of the glycan and for the mass change resulting from the reductive amination. The mass calculations are shown in the following example of the derivatisation of N-acetylglucosamine with 2-aminopyridine (PA).
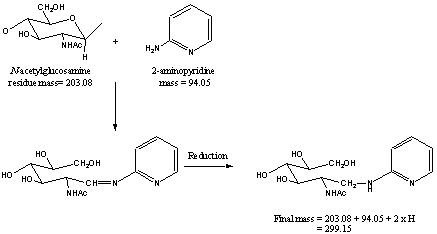
Derivisation of N-acetylglucosamine with 2-aminopyridine (PA).
If the glycans have been permethylated or peracetlyated this is selected when choosing the nature of the monosaccharides that may be present in the composition (see below).
-
-
Monosaccharide residues
GlycoMod is designed to calculate the masses of oligosaccharides using underivatized, permethylated or peracetylated monosaccharides since mass spectrometric data is often obtained from these later derivatized oligosaccharides.
The user may stipulate which monosaccharides are/are not/or may possibly be present in your glycan. You may also enter a range of values. For example, from monosaccharide analysis the user may know that the glycan contains fucose and since it is an N-linked glycan released using PNGase it must contain N-acetylglucosamine and mannose.
Since it is often possible to obtain the same mass arising from several monosaccharide compositions, the more information entered regarding which monosaccharide residues are, or are not, present will mean that fewer misleading compositions are returned.
There are some pre-programmed limits to the possible compositions allowed for N-linked glycans. These were implemented after careful investigation of the known N-linked glycan structures.
- A composition may not contain both sulfate and phosphate.
- The sum of the number of hexose plus HexNAc residues must be greater than or equal to the number of sulfate or phosphate residues.
- The sum of the number of hexose plus HexNAc residues cannot be zero, i.e., an N-linked glycan contain either a hexose or a HexNAc residue, or both.
- The number of fucose residues plus 1 must be less than or equal to the sum of the number of hexose plus HexNAc residues.
- If the number of HexNAc residues is less than or equal to 2 and the number of hexose residues is greater than 2, then the number of NeuAc and NeuGc residues must be zero.
There are no pre-programmed limits to the possible compostitions allowed for O-linked glycans, except for the total number of any one particular monosaccharide residue.
The total number of individual monosaccharides is limited for both N-linked and O-linked oligosaccharides. These limits are listed below and have been set by analysing the literature.
N-linked oligosaccharides O-linked oligosaccharides Hexose 0-20 0-14 HexNAc 0-20 0-14 Deoxyhexose 0-6 0-6 NeuAc 0-5 0-7 NeuGc 0-3 0-7 Pentose 0-4 0-3 Sulphate 0-3 0-6 Phosphate 0-2 0-6 KDN - 0-2 HexA - 0-2
KDN and HexA are not allowed for N-linked oligosaccharides as these residues have only been found on O-linked oligosaccharides so far.
An upper limit on the total mass of the glycoform has been set. This limit is 8000 Da for underivatized, 10000 Da for permethylated and 13000 Da for peracetylated N-linked glycans. For O-linked glycans the limit is 5000 Da for underivatized, 7000 Da for permetylated and 9500 Da for peracetylated oligosaccharides.
Output parameters
The output for GlycoMod is divided into two main sections - a header and a table for each user mass entered.
The header section lists the monosaccharide compositional data entered by the user and the calculated peptide masses of a protein sequence or UniProtKB AC or ID if “Glycopeptide” was chosen.
The output tables report the monosaccharide compositions whose theoretical masses match the entered experimental user mass after any stated derivative or peptide modification. A separate table is generated for each entered mass. Each table shows the glycoform mass, Δmass in daltons or ppm (depending on units entered by the user on the input form), the monosaccharide composition, and the glycan type if N-linked (see below).
The structure of N-linked glycans is generally well conserved with a core region consisting of 2 N-acetylglucosamine residues and 3 mannose residues (Man3GlcNAc2), and branches containing a variety of hexose and HexNAc residues that may be further substitued with other residues such as sialic acid (see figure below). To help the user to distinguish between those residues residing in the core of the glycan and those on the branches, when the composition contains at least 2 HexNAc residues and 3 Hexose residues these are removed from the overall composition and written separately, e.g., (Hex)2(HexNAc)3(Deoxyhexose)1 + (Man)3(GlcNAc)2.
The glycan type of N-linked glycans is also given, i.e., hybrid/complex or high mannose (see figure below). These are classified by:
- If the number of HexNAc residues equals 2 and the number of hexose residues is greater than or equal to 5, then the N-linked glycan is of the type “high mannose”.
- If the number of HexNAc residues is greater than or equal to 3 and the number of hexose residues is also greater than or equal to 3, then the N-linked glycan is of the type “hybrid/complex”.
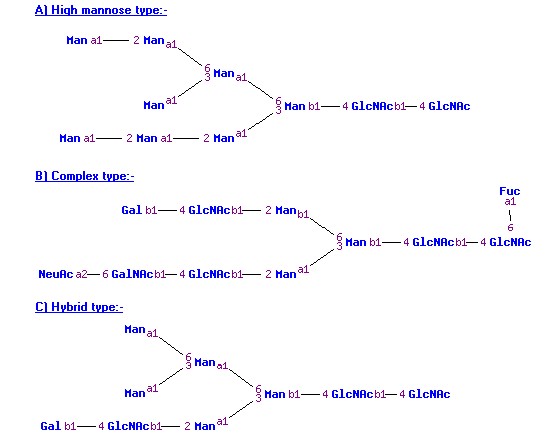
Classification of N-linked glycan structures
There are no defined glycan types for O-linked glycans in GlycoMod.
If a glycopeptide mass is entered together with a protein sequence or UniProtKB AC or ID, then GlycoMod calculates the possible oligosaccharide compositions attached to the unmodified theoretical peptides formed after enzymatic or chemical digestion. GlycoMod also considers the peptides that may be biologically modified (as annotated in UniProtKB) and/or chemically modified (as specified by the user in the input form). If the entry was a UniProtKB, the description line from the UniProtKB entry and link to the SWISS-PROT/TrEMBL entry is given.
When a glycopeptide mass is entered, each table contains additional information on the peptide mass [M], peptide sequence or a UniProtKB AC or ID (where entered by the user), the theoretical glycopeptide mass, and any post-translational modification annotated in UniProtKB if a UniProtKB AC or ID was entered.
If GlycoMod suggests a composition that has been reported in the GlyConnect database of glycan structures, a link to the corresponding GlyConnect entry is provided. The user can also select to display compositions reported in GlyConnect separately from the compositions not known in the database.